
Nesta sessão será apresentado um fluxo de análise, a partir de um conjunto de dados de amplicons do gene 16S rRNA. Os dados estarão disponíveis somente durante o minicurso da Escola Paranaense de Bioinformática 2024 (06 a 08 de novembro, Londrina – PR). Entretanto, a sequência de comandos de toda análise pode ser adaptada a outros conjuntos de dados.
O conjunto de dados que será usado nesta atividade é composto por sequências amplicon do gene 16S rRNA de bactérias e arquéias, obtidas a partir de amplificação por PCR e sequenciamento NGS de DNA metagenômico obtido de fezes de camundongo. Será usado um conjunto de dados reduzido para facilitar a execução das análises e a interpretação dos resultados. DNA metagenômico foi obtido de dois grupos de camundongos (com três réplicas por tratamento): (i) grupo controle, com dieta normal (Contol) e (ii) grupo tratado com uma dieta hipercalórica (HighDiet). Após 20 semanas, observou-se grande diferença na massa corporal dos animais submetidos às duas diferentes dietas (Figura 2), quando então as fezes foram amostradas e o DNA metagenômico extraído.
A amplificação do gene 16S rRNA foi realizada usando os iniciadores 515F/806R, que cobre a região V4. O sequenciamento dos amplicons foi realizado em plataforma Illumina MiSeq, usando a estratégia paired-end. Verifique detalhes sobre os iniciadores no sítio do Earth Microbiome Project (EMP) . Use estas informações e a ferramenta Web BLAST para responder as questões abaixo.
Q. Quais as sequências de bases dos iniciadores direto e reverso?
Q. Qual a região de anelamento dos iniciadores na sequência do gene 16S rRNA?
Q. Qual o tamanho aproximado esperado para o amplicon?
Q. Qual a sobreposição esperada para os pares de sequências?

As amostras estão listadas abaixo e tetalhes para as leituras de sequências obtidas podem ser visualizados nos links correspondentes.
A execução de todos os comandos nesta atividade é realizada a partir do diretório de análise ("analysis"), exceto quando indicado de forma diferente.
Inicialmente, é necessário criar os diretórios para armazenar os arquivos do conjunto de dados e, posteriormente, arquivos que serão gerados durante a análise.
# Criar os diretórios da forma indicada abaixo
# activity
# ├── analysis
# │ ├── dada2
# │ ├── diff
# │ ├── div
# │ ├── phylo
# │ └── tax
# ├── data
# └── db
# No Linux, estes diretórios podem ser criados com o comando abaixo
$ mkdir -p data db analysis/{dada2,tax,phylo,div,diff}
Baixe e transfira todos os arquivos "fastq" (somente estes arquivos) das amostras para o diretório "data" e baixe e transfira o arquivo "map.tsv" para o diretório "analysis".
Esta atividade será executada com o programa qiime2. O programa é multiplataforma e pode ser instalado e executado de diferentes formas. Detalhes sobre a instalação podem ser verificados na documentação do programa . Aqui, toda a análise é executada na linha de comando de um terminal em ambiente Linux. A atividade será executada de forma demostrativa e os resultados de cada etapa estão disponíveis para serem baixados. Estes arquivos possuem extensão ".qzv" e podem ser visualizados através de um navegador, acessando o endereço view.qiime2.org .
Em um terminal, acesse o diretório de análise e inicie o programa qiime2.
# Iniciar o qiime2
# Atenção! A versão do programa pode ser diferente
$ conda activate qiime2-2024.5
# Tabulando os metadados
$ qiime metadata tabulate \
--m-input-file map.tsv \
--o-visualization map.qzv
Nesta atividade serão usados dados de leituras de sequências gerados na plataforma Illumina MiSeq, que já passaram pelo processo de desmultiplexação, ou seja, cada sequência já foi associada a uma amostra. Arquivos contendo as leituras de sequências correspondentes a cada uma das amostras foram gerados no processo de desmultiplexação. As leituras de sequências nestes arquivos estão no formato conhecido como Casava 1.8 Fastq . O diretório "data" contém arquivos compactados no formato "fastq", separados por amostra, sendo que cada amostra possui dois arquivos, identificados como "R1" e "R2", indicando que o sequenciamento foi realizado usando a estratégia "paired-end". O nome dos arquivos está no formato "IN9_8_L001_R1_001.fastq.gz", onde cada identificador, separado pelo caractere "_", indica, na sequência: (i) identificador da amostra, (ii) sequência do barcode, (iii) lane, (iv) direção do sequenciamento e (v) número de série. A extensão ".gz" indica que o arquivo está no formato compactado "gzip e são usados desta forma na análise.
Antes de iniciar a análise é necessário importar todos dados de sequências, salvos no diretório "data", para um arquivo do tipo artefato do programa qiime2.
# Importando os dados de sequências para um artefato ".qza"
$ qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path ../data \
--input-format CasavaOneEightSingleLanePerSampleDirFmt \
--output-path demux.qza
Após a importação dos dados, é útil gerar um resumo que permitirá determinar quantas sequências foram obtidas por amostra e também obter um resumo da distribuição das qualidades das sequências para cada posição. O artefato com os dados foi gerado dentro do diretório de análise. Toda a execução de comandos, deste ponto em diante, ocorrerá a partir deste diretório de análise como referêcia.
# Gerando um resumo dos dados
$ qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
O plugin dada2 pode ser usado para detectar e corrigir (quando possível) os dados de sequências amplicons gerando na plataforma Illumina, remover a redundância nas sequências, filtrar sequências quiméricas e também montar os pares de sequências (caso o sequenciamento tenha sido realizado com bibliotecas do tipo "paired-end"). O resultado será os artefatos: (i) "FeatureTable[Frequency]", contendo contagem (frequências) de cada sequência única (ASV) em cada uma das amostras e (ii) "FeatureData[Sequence]", mapeia os identificadores de características na "FeatureTable" para as sequências que eles representam.
OBS. O método usado requer dois parâmetros que são usados no filtro de qualidade: (i) "--p-trim-left m", poda as primeiras "m" bases de cada sequência e (ii) "--p-trunc-len n", trunca cada sequência na posição "n", permitindo remover regiões de baixa qualidade nas sequências (para determinar quais valores devem ser passados a estes dois parâmetros, deve-se rever o resumo da importação de dados mostrado acima). Os sufixos "f" e "r" nestes parâmetros referem-se às sequências diretas (forward, do arquivo R1) e reversas (reverse, do arquivo R2). O comando para sequências do tipo "paired-end" é mostrado abaixo.
# Realizando a etapa de denoising com o método dada2
$ qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux.qza \
--p-trim-left-f 0 \
--p-trunc-len-f 247 \
--p-trim-left-r 0 \
--p-trunc-len-r 175 \
--p-n-threads 3 \
--o-representative-sequences dada2/rep-d2-demux.qza \
--o-table dada2/tbl-d2-demux.qza \
--o-denoising-stats dada2/sts-d2-demux.qza
Em seguida, uma visualização pode ser gerada com o comando abaixo.
# Visualização para as estatísticas da análise
$ qiime metadata tabulate \
--m-input-file dada2/sts-d2-demux.qza \
--o-visualization dada2/sts-d2-demux.qzv
# Visualização para a tabela de frequências por amostra
$ qiime feature-table summarize \
--i-table dada2/tbl-d2-demux.qza \
--m-sample-metadata-file map.tsv \
--o-visualization dada2/tbl-d2-demux.qzv
# Visualização para lista de sequências representativas (ASV)
$ qiime feature-table tabulate-seqs \
--i-data dada2/rep-d2-demux.qza \
--o-visualization dada2/rep-d2-demux.qzv
sts-d2-demux.qzv
tbl-d2-demux.qzv
rep-d2-demux.qzv
A partir dos resultados no arquivo "sts-d2-demux.qzv", responda.
Q. Qual o número médio de leituras de sequências por amostra? Quais amostras contém o menor e o maior número de leituras de sequências? Que porcentagens do número inicial de leituras de sequências representam?
Q. Qual a etapa crítica no processo de filtragem? Aquela que mais reduziu o número de leituras de sequências nas amostras.
A partir dos resultados no arquivo "tbl-d2-demux.qzv", responda.
Na aba "Overview"...
Q. Qual o número total de amostras, ASV e leituras de sequências?
Q. Qual a média de leituras de sequências por amostra
Q. Qual a média de leituras de sequências por ASV?
Na aba "Interactive Sample Detail"...
Q. Identifique quais amostras contém o menor e maior número de leituras de sequências. A quais grupos pertencem?
Q. No caso de aplicarmos um limite mínimo de leituras de sequências por amostra, quantas e de quais grupos seriam perdidas para: (a) 5.000; (b) 10.000?
Na aba "Feature Detail"...
Q. Identifique a sequência ASV mais frequente (mais abundante). Em quantas amostras esta sequências ocorre?
A partir dos resultados no arquivo "rep-d2-demux.qzv", responda.
Q. Quais os valores mínimo, máximo e médio para os comprimentos das leituras de sequênicas?
Q. Estes valores estão dentro do esperado para os amplicons do gene 16S rRNA gerados neste trabalho?
Q. Interprete os percentis para os comprimentos das leituras de sequências. Estão dentro do esperado?
Q. Agora observe a primeira ASV listada. O que representa o seu "Feature ID"? Faça a identificação taxonômica para esta ASV usando o programa BLAST (a sequência de DNA contém um link para o programa). Qual a frequência e a distribuição desta ASV entre as amostras (verificar no arquivo "tbl-d2-demux.qzv")?
Neste ponto é possível explorar a composição taxonômica das amostras e relacioná-la aos metadados. O primeiro passo é identificar taxonomicamente as sequências representativas (ASV) comparando-as contra um banco de dados contendo sequências referência com classificação taxonômica conhecida. Aqui é importante notar que existem diferentes opções de programas e bancos de dados, que irão gerar resultados diferentes. Além disso, pode-se aplicar estratégias diferentes usando um mesmo programa.
Resumidamente, o qiime2 disponiviliza três alternativas para a classificação taxonômica: (i) BLAST, (ii) VSEARCH e (iii) classify-sklearn. A opção "classify-sklearn" usa um classificador "Naive Bayes" pré-treinado a partir de sequências referências em um banco de dados.
Nesta atividade, será usado um classificador já pré-treinado com o banco de dados Greengenes 13_8 99% OTUs. Inicialmente, as sequências no banco de dados foram agrupadas, em OTU, com um limite de identidade de 99%. Em seguida, somente as cerca de 250 bases da região V4 do gene 16S rRNA (correspondente à região de amplicon obtida na PCR com o par de iniciadores 515F/806R) foram extraídas e mantidas no banco de dados. Finalmente, este banco de dados foi utilizado para treinamento do classificador.
Baixe o arquivo gerado para o classificador (gg-13-8-99-515-806-nb-classifier.qza ) e salve no diretório "db"
O arquivo do classificador pode, então, ser usado para identificação taxonômica das ASV com o comando abaixo.
# Classificação taxonômica das ASV
$ qiime feature-classifier classify-sklearn \
--i-classifier ../db/gg-13-8-99-515-806-nb-classifier.qza \
--i-reads dada2/rep-d2-demux.qza \
--p-n-jobs 3 \
--o-classification tax/tax-rep-d2-demux.qza
A partir do arquivo contendo as ASV classificadas taxonomicamente, gerado na análise anterior, um gráfico de distribuição taxonômica, em vários níveis e interativo, pode ser gerado.
# Gerar o gráfico de barras
$ qiime taxa barplot \
--i-table dada2/tbl-d2-demux.qza \
--i-taxonomy tax/tax-rep-d2-demux.qza \
--m-metadata-file map.tsv \
--o-visualization tax/tbl-d2-demux_barplot.qzv
A classificação taxonômica pode ser unida com as sequências ASV.
# Unindo sequências e taxonomia
$ qiime metadata tabulate \
--m-input-file dada2/rep-d2-demux.qza \
--m-input-file tax/tax-rep-d2-demux.qza \
--o-visualization tax/taxASV-rep-d2-demux.qzv
A classificação taxonômica pode ser filtrada para qualquer taxa. No exemplo abaixo, são extraídas as classificações para o filo Firmicutes, que é o mais abundante nas amostras.
# Aplica o filtro para Firmicutes
# Use "p_" para manter somente as características com identificação mínima de filo
$ qiime taxa filter-table \
--i-table dada2/tbl-d2-demux.qza \
--i-taxonomy tax/tax-rep-d2-demux.qza \
--p-include Firmicutes \
--o-filtered-table tax/tbl-d2-demux_Firmicutes.qza
# Gera um resumo para ser visualizado
$ qiime feature-table summarize \
--i-table tax/tbl-d2-demux_Firmicutes.qza \
--m-sample-metadata-file map.tsv \
--o-visualization tax/tbl-d2-demux_tblFirmicutes.qzv
# Gera uma tabela de frequências por amostra
$ qiime metadata tabulate \
--m-input-file tax/tbl-d2-demux_Firmicutes.qza \
--o-visualization tax/tbl-d2-demux_freqFirmicutes.qzv
tbl-d2-demux_tblFirmicutes.qzv
tbl-d2-demux_freqFirmicutes.qzv
Uma árvore filogenética precisa ser criada para análises posteriores, como cálculo de alguns índices de diversidade (ex., Faith's phylogenetic diversity e UniFrac). OBS.: Os resultados gerados para esta etapa da análise serão gravados no diretório "phylo".
# Primeiro, gerar um alinhamento
$ qiime alignment mafft \
--i-sequences dada2/rep-d2-demux.qza \
--o-alignment phylo/aln-rep-d2-demux.qza
# Remove posições muito variáveis no alinhamento
$ qiime alignment mask \
--i-alignment phylo/aln-rep-d2-demux.qza \
--o-masked-alignment phylo/msk-aln-rep-d2-demux.qza
# gera árvore sem raiz (ut)
$ qiime phylogeny fasttree \
--i-alignment phylo/msk-aln-rep-d2-demux.qza \
--o-tree phylo/ut-msk-aln-rep-d2-demux.qza
# inclui raiz na arvore (rt)
$ qiime phylogeny midpoint-root \
--i-tree phylo/ut-msk-aln-rep-d2-demux.qza \
--o-rooted-tree phylo/rt-msk-aln-rep-d2-demux.qza
msk-aln-rep-d2-demux.qza
msk-aln-rep-d2-demux.qza
rt-msk-aln-rep-d2-demux.qza
A árvore filogenética gerada pode ser visualizada com a ferramenta on line iTOL .
É importante notar que, para estas análises, é necessário ter as leituras de sequências agrupadas por ASV ou OTU e as frequências destas ASV/OTU por amostra, mas não é necessária a classificação taxonômica. Inúmeras métricas de diversidade estão disponíveis no programa e aquelas calculadas por padrão, são:
As análises de diversidade no qiime2 estão disponíveis através do plugin "q2-diversity", que permite gerar índices de alfa e beta diversidade para as amostras e aplicar testes estatísticos relacionados.
Inicialmente as amostras são submetidas a rarefação com uma resolução (ou profundidade) definida pelo usuário. Isso normalizará o número de leituras de sequências por amostra (todas as amostras conterão o mesmo número de sequências). Em seguida, as métricas de alfa e beta diversidade são calculadas para as amostras rarefeitas.
O valor limite para o número de leituras de sequência por amostra na rarefação é definido no parâmetro "--p-sampling-depth". As leituras de sequências em cada amostra serão aleatoriamente selecionadas, sem reposição, até atingir o valor de rarefação definido. Caso a amostra contenha um número de leituras de sequências inferior ao definido para a rarefação, a amostra será descartada da análise.
A tabulação pode ser combinada com os metadados e entre várias métrica e outras informações.
# Cálculo das métricas de diversidade
$ qiime diversity core-metrics-phylogenetic \
--i-table dada2/tbl-d2-demux.qza \
--i-phylogeny phylo/rt-msk-aln-rep-d2-demux.qza \
--p-sampling-depth 10000 \
--m-metadata-file map.tsv \
--output-dir core-metrics
# Tabulando as métricas de alfa diversidade
# É possível também incluir no comando os metadados com a opção "--m-input-file map.tsv"
$ qiime metadata tabulate \
--m-input-file core-metrics/observed_features_vector.qza \
--m-input-file core-metrics/evenness_vector.qza \
--m-input-file core-metrics/shannon_vector.qza \
--m-input-file core-metrics/faith_pd_vector.qza \
--o-visualization core-metrics/alpha_diversity_merged.qzv
# Tabulando gráfico PCoA com alfa diversidade
$ qiime emperor plot \
--i-pcoa core-metrics/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file map.tsv \
--m-metadata-file core-metrics/faith_pd_vector.qza \
--o-visualization core-metrics/unweighted_unifrac_pcoa_results_with_faith_pd.qzv
Alfa diversidade
alpha_diversity_merged_merged.qzv
Beta diversidade
jaccard_emperor.qzv
bray_curtis_emperor.qzv
weighted_unifrac_emperor.qzv
unweighted_unifrac_emperor.qzv
unweighted_unifrac_pcoa_results_with_faith_pd.qzv
Um diretório "core-metrics" será criado, contendo diversos arquivos gerados na análise. Um resumo da amostras rarefeitas pode ser gerado para visualização com o comando abaixo. Compare este resumo com aquele gerado para as amostras não rarefeitas.
# Resumo da distribuição de ASV por amostra rarefeita
$ qiime feature-table summarize \
--i-table core-metrics/rarefied_table.qza \
--m-sample-metadata-file map.tsv \
--o-visualization core-metrics/rarefied_table.qzv
É possível criar um gráfico de rarefação alfa, onde uma métrica de alfa diversidade é calculada para a rarefação das amostras em valores crescentes. Um valor para rarefação é definido, uma sub-amostra aleatória das leituras de sequência é gerada para cada amostra e uma métrica de alfa diversidade é calculada para as sub-amostras. Um novo valor (maior) de rarefação é então definido, as sub-amostras re-criadas e a métrica re-calculada. Novos valores de rarefação são definidos até atingir o número máximo de leituras de sequências nas amostras.
No comando abaixo, o parâmetro "--p-max-depth" irá controlar a rarefação máxima atingida na análise. O parâmetro "--p-steps" define o número de rarefações que serão realizadas. Por exemplo, se o parâmetro "--p-max-depth" for definido como "10000" e o parâmetro "--p-steps" como "10", então cada rarefação será realizada com valores adicionais de 1.000 leituras de sequências (10.000 / 10 = 1.000), ou seja, os pontos de rarefação serão: 1.000, 2.000, 3.000,...,10.000. Por outro lado, se o parâmetro "--p-steps" for definido como "20", serão realizados 20 sub-amostragens com valores adicionais de 500 (10.000 / 20 = 500).
# Gráfico de rarefação alfa
$ qiime diversity alpha-rarefaction \
--i-table dada2/tbl-d2-demux.qza \
--i-phylogeny phylo/rt-msk-aln-rep-d2-demux.qza \
--p-max-depth 10000 \
--p-steps 10 \
--m-metadata-file map.tsv \
--o-visualization div/raref10k-tbl-d2-demux.qzv
raref10k-tbl-d2-demux.qzv (resultado para análise com --p-step 10)
raref10k20s-tbl-d2-demux.qzv (resultado para análise com --p-step 20)
O resultado irá mostrar dois gráficos. O gráfico, no topo, é o gráfico de rarefação alfa e é basicamente usado para determinar se a riqueza das amostras foi completamente observada ou sequenciada. Se o gráfico tende a um ponto de saturação, realizar a rarefação com valores maior a partir deste ponto, não irá acrescentar diversidade às amostras, ou seja, a amostragem é representativa e suficiente para a diversidade do ambiente estudado. Se as linhas ainda mostram uma tendência de aumento na métrica de diversidade, indica que a diversidade nas amostras não foi, ainda, completamente observada (porque poucas sequências foram coletadas). O gráfico na parte inferior é importante para agrupar amostras por metadados. O gráfico ilustra o número de amostras que permanecem em cada grupo ao longo das rarefações.
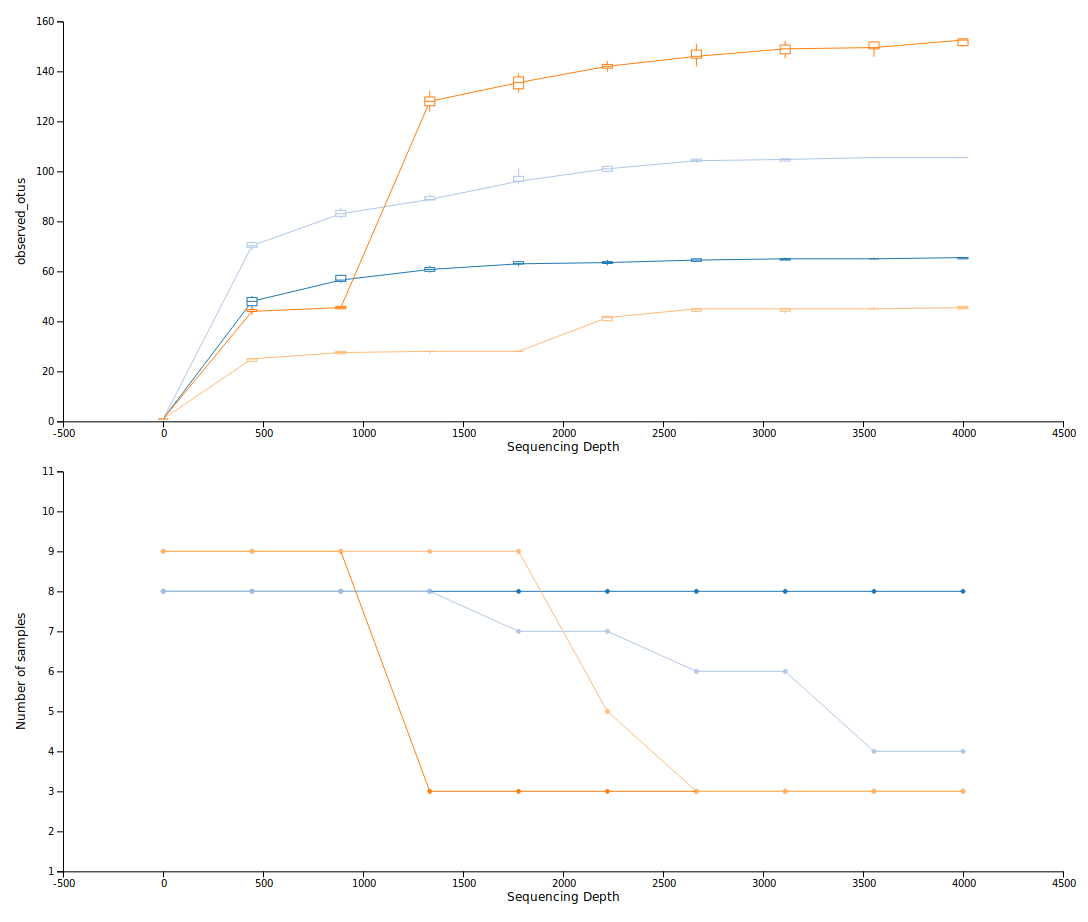
A figura mostra os dois gráficos gerados na análise de rarefação alfa para um conjunto de amostras que foram agrupadas por tratamento. As linhas em cores diferentes representam os grupos de amostras para cada tratamento. O gráfico na parte superior é a rarefação alfa, mostrando que houve saturação na alfa diversidade (neste caso, para a riqueza) para todos os tratamentos. O gráfico na parte inferior da figura mostra que até o ponto de rarefação de cerca de 800 leituras de sequências por amostra, nove amostras são usadas para calcular a métrica de diversidade no grupo representado pela linha laranja. Mas, a partir deste ponto, somente três amostras estão incluídas no grupo (isso porque as demais seis amostras não possuem número suficiente de leituras de sequências). Neste mesmo ponto é observado um aumento de riqueza para este tratamento no gráfico de rarefação alfa, mas que indica ser uma tendência devido a perda de amostras.
No próximo passo, é possível testar as associações entre a diversidade alfa e diferentes dados categóricos presentes nos metadados (fazendo as comparações entre tratamentos), gerar gráficos do tipo box plot e avaliar a significância para as alfa diversidades calculadas, aplicando testes estatísticos não paramétricos. Aqui é aplicado o teste de Kruskal-Wallis (teste H), um teste não paramétrico que compara as medianas entre dois ou mais grupos (considerado uma alternativa não paramétrica a ANOVA). O comando abaixo testa essa associação em relação a métrica de "faith_pd", mas ela pode ser testada para qualquer outra métrica de alfa diversidade (arquivos contendo "vector" no nome).
# Teste de significância não paramétrico para alfa diversidade
$ qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics/faith_pd_vector.qza \
--m-metadata-file map.tsv \
--o-visualization core-metrics/faith_pd_group_significance.qzv
observed_features_group_significance.qzv
evenness_group_significance.qzv
shannon_group_significance.qzv
faith_pd_group_significance.qzv
Os resultados irão mostrar se existe alguma diferença estatística significativa entre os tratamentos, quando comparados por uma das variáveis categóricas presentes nos metadados. Por exemplo, nesta atividade, as amostras podem ser divididas em dois tratamentos: (i) Control (controle) e (ii) HighDiet (dieta calórica), através da sétima coluna do arquivo de metadados. Nos resultados são apresentados o valor do teste estatístico H, o valor-p e o valor-q (que representa o valor-p corrigido para multiplas comparações).
Q. Verifique se há significancia na comparação entre os tratamentos.
Q. Por que, neste caso, o valor-p e o valor-q são iguais?
As métricas de beta diversidade, automaticamente geram gráficos baseados em PCoA (análise de coordenadas principais; arquivos contendo "emperor" no nome).
Q. Visualize o resultado para análise PCoA com a métrica Bray-Curtis e identifique as amostras por tratamento.
Q. Existe uma tendência para separação das amostras por tratamento?
Q. Qual a porcentagem de variação nos dados explicada pelos três eixos da análise?
Q. Esta tendência é também observada em outras métricas de beta diversidade?
É possível ainda aplicar testes estatísticos de hipótese para verificar se há diferença significativa entre tratamentos (diferenças que podem, por exemplo ser observadas qualitativamente nos gráficos PCoA). Esta versão da análise aplica o método PERMANOVA, um teste estatístico de hipóteses multivariado não paramétrico, que compara grupos baseado em medidas de distâncias. Semelhante ao teste de ANOVA, aplicado a variáveis paramétricas, fazendo uso do teste F para comparar a variância dentro do grupo com a variância entre grupos. Entretanto, o teste não presupõem a distribuição normal dos dados e realiza testes de significância comparando o resultado real do teste F com um pseudo testes F, obtidos a partir de permutações aleatórias dos objetos entre os grupos. A análise irá testar se a distância entre amostras dentro de um mesmo grupo são mais similares entre si do que suas similaridades com amostras em grupos diferentes.
Q. Qual o número de permutações usadas neste teste?
Q. Há diferenças significativas entre as amostras?
# Teste para beta diversidade entre os tratamentos
# Obs. os tratamentos comparados são estabelecidos na opção "--m-metadata-column"
$ qiime diversity beta-group-significance \
--i-distance-matrix core-metrics/bray_curtis_distance_matrix.qza \
--m-metadata-file map.tsv \
--m-metadata-column Treatment \
--o-visualization core-metrics/bray_curtis_treatment_significance.qzv
jaccard_treatment_significance.qzv
bray_curtis_treatment_significance.qzv
unweighted_unifrac_treatment_significance.qzv
weighted_unifrac_treatment_significance.qzv
As diferenças nas estruturas das comunidades microbianas em amostras ambientais pode ocorrer devido a diferenças na presença/ausência de grupos taxonômicos/fiologenéticos ou devido a diferenças na sua abundância. No segundo caso, o teste ANCOM-BC pode ser aplicado para identificar características (por ex., ASV) que apresentam diferença na abundância entre os grupos de amostras (por ex., entre tratamentos). O teste aplica um modelo de regressão linear com reconhecimento de composição, aplicando correção de viés.
O teste é aplicado para comparar dois grupos de amostras somente e caso haja mais de dois tratamentos para a comparação, um filtro deve ser aplicado, antes de executar a análise, para extrair apenas dois grupos de amostras a serem comparadas. Os grupos de amostras são identificados a partir do arquivo de metadados. Posteriormente, a análise é aplicada às amostras filtradas. Nesta atividade há apenas dois grupos de amostras, separadas pelo tratamento da dieta (controle e dieta calórica), não sendo necessário a aplicação de filtro inicial.
# Análise de abundância diferencial
$ qiime composition ancombc \
--i-table dada2/tbl-d2-demux.qza \
--m-metadata-file map.tsv \
--p-formula 'Treatment' \
--o-differentials diff/ancombc-treatment.qza
# Criação de gráfico barplot com os resultados
$ qiime composition da-barplot \
--i-data diff/ancombc-treatment.qza \
--p-significance-threshold 0.001 \
--o-visualization diff/da-barplot-treatment.qzv
O gráfico barplot gerado mostra as ASV que apresentam abundância diferencial entre os grupos de amostra controle e dieta calórica, indicando em qual tratamento cada ASV está enriquecida.
Q. Quantas ASV são diferencialmente expressas?
Q. Quais ASV são mais e menos expressas no tratamento de dieta calórica?
Q. Quais são os grupos taxonômicos diferencialmente expressos? Estão proximamente relacionados?
Última atualização: 29 de outubro de 2024